はじめまして. 大阪大学蛋白質研究所細胞システム研究室修士1年の道田大貴と申します. この記事では近年急速に発達している分子生物学実験技術と計算機技術を組み合わせる技術であるバイオインフォマティクスについてお話させていただこうと思います.
目次
そもそも, 生体内はどんなふうに制御されているの?
DNA, RNA, 蛋白質
みなさんはおそらく「遺伝子」という言葉を聞いたことがあると思います。遺伝子とは細胞の核の中にあるDNAという分子に書かれている暗号です.
この暗号が一旦mRNAと呼ばれる分子にコピーされたあと(転写)それを解読すること(翻訳)によって蛋白質が作られます.

蛋白質は非常に様々な種類がありそれぞれが独自のはたらきを持っています. 例えばみなさんもよく耳にする酵素と呼ばれる蛋白質は生体内で様々な化学反応を引き起こします.
生物の個体ごとの違いはそれぞれが持つ遺伝子が異なっていることに由来します. 遺伝子が異なればからだの中で作られる蛋白質の種類, 量, 機能が異なるからです.
たとえば日本人などは目に黒い色素蛋白を多く持っているので目が黒いですが, 西洋人などはそれの量が少ないので目が青くなっています. 目の色素蛋白の情報が書いてある遺伝子が異なるからです.
つまり蛋白質は生体の外見, 病気の有無, 性格などを決定する非常に大事なものなのです.
生体内はまるでオーケストラ!
生体内では実にたくさんの蛋白質が相互作用しながらからだ全体を調節しています. これにはそれぞれの蛋白質の活性の強さのバランスや, はたらくタイミングが適切に制御されることが重要です.

たとえば細胞分裂を行う必要がないときに細胞分裂を促進する蛋白がずっと活性化していては困ります. 異常に増殖した細胞はいつかからだを蝕みます. がんはこのような仕組みで起こる病気です.
生体内の蛋白質の相互作用はまるでオーケストラの演奏のようです. オーケストラで良い演奏を行うにはそれぞれの楽器の音量がちょうどよく調節することや, 適切なリズム, つまり適切なタイミングで音を出すことが不可欠です.
楽譜にはどのような音量で演奏するか, どのようなタイミングで音を出すかが書かれています. 生体内ではこの楽譜の役割を遺伝子が担っています.
ところが楽譜に音だけが書いてあっても音量などの表現の仕方がわからないのと同様に遺伝子に蛋白質の構造の情報のみが書かれているだけではこのような複雑な制御は不可能です.
楽譜にはフォルテ、ピアノといった音量の指示やespressivo(表情豊かに)というようにイタリア語や英語で演奏表現の指示が書いてあるのですが遺伝子にも「エピジェネティック制御」と呼ばれるそれと似ている仕組みがあります.
DNAはヒストンと呼ばれる蛋白質に電化製品のコードのように巻き付いています. エピジェネティック制御とはこのヒストンに化学反応によってマークがつけられたり, 蛋白質が直接DNAに結合するなどの制御のことです. これにより遺伝子が転写される量やタイミングが制御されます.
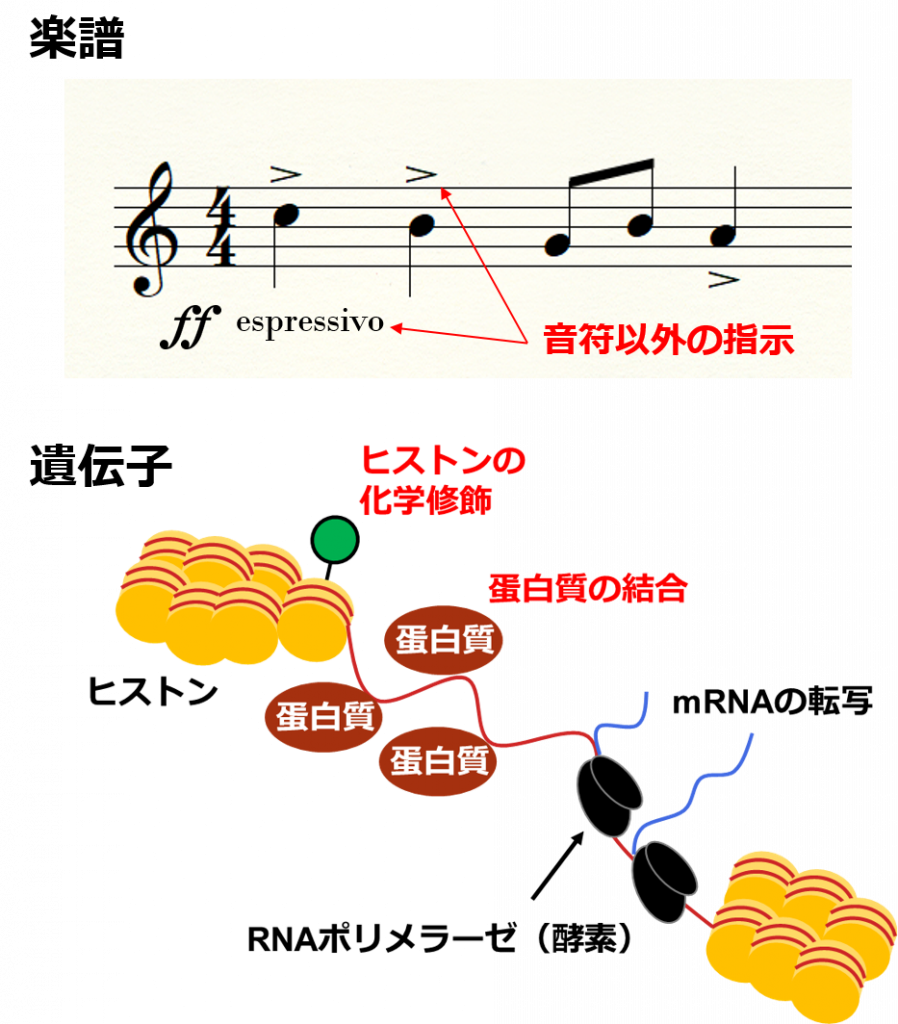
エピジェネティック制御が遺伝子発現の振る舞いを確実に制御することで私達のからだは一流オーケストラのように的確に調節されているわけです.
次の章ではまずエピジェネティック制御を含めた遺伝子に関するデータを網羅的に取得することのできる次世代シークエンサーについてお話します.
〇〇-seqで調べるDNAの様子
人類の秘密兵器, 次世代シークエンサー!
生きていくために必要な遺伝子の1セットのことをゲノムといいます. 遺伝子はDNAを構成する4種類の塩基 (A,T,G,C)によって暗号化されて書かれています. ヒトのゲノムは約30億塩基で書かれていることがわかっています.
昔は一塩基ずつ人が読み取っていたので一人で30億塩基も読むなんてことは不可能でした. ところが近年開発されて今も急速に発展しつつある次世代シークエンサーという技術を使うとなんと3日以内に30億塩基読むことができてしまいます.
この次世代シークエンサーを使用することでゲノム全体のDNAの様子を網羅的に調べることができます.
ChIP-seq
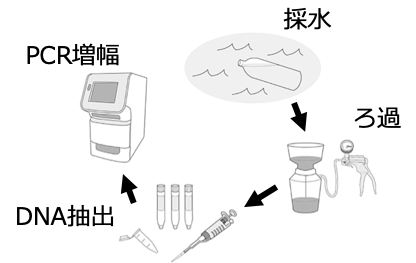
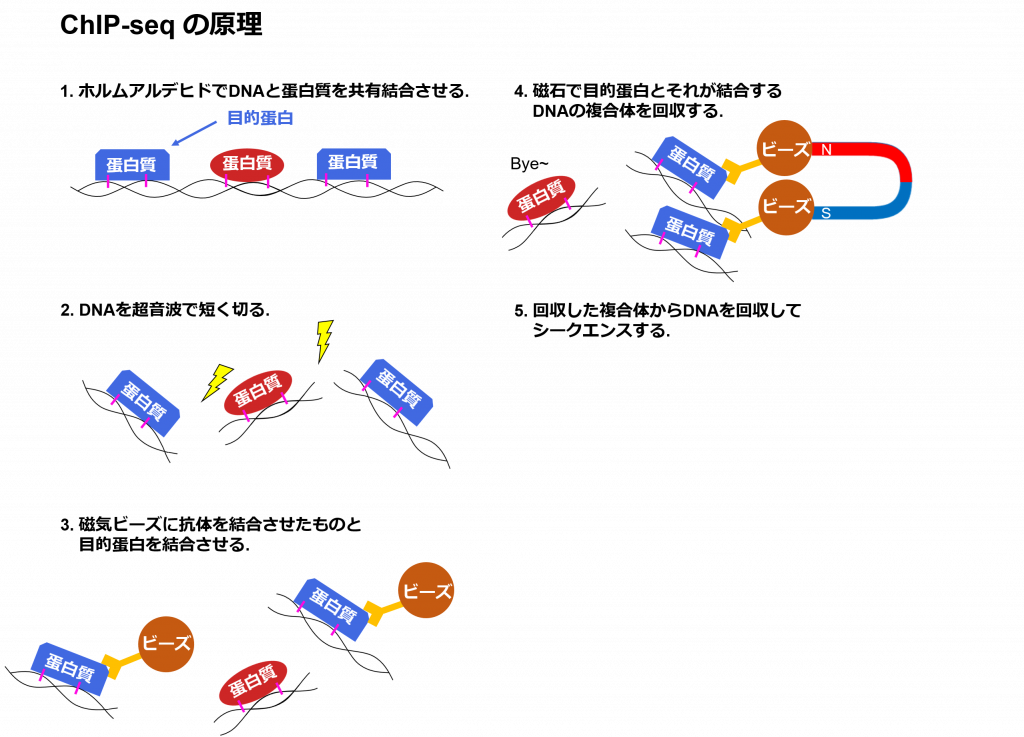
まずは次世代シークエンサーを使ってゲノムのどの領域にヒストンマークや蛋白質の結合が起こっているかを調べるChIP-seq (チップセック) についてお話します.
細胞からDNAを回収して超音波で短く断片化します. そのあと抗体を使って目的のヒストンマーカーや蛋白質が結合しているDNAのみを回収します. これを次世代シークエンサーで読むことで目的のヒストンマーカーや蛋白質が結合していたDNAの配列を取得できます.
ちなみに私は主にこのChIP-seqを実験から解析まですべて行っていますがこの実験は非常に難しいです.
ATAC-seq
蛋白質がDNAに結合するにはDNAがヒストンに巻き付いた状態ではなく, 解けていなければなりません. 逆にDNAが解けているところはエピジェネティック制御が起きていたり遺伝子が活性化している場所であると考えられます.
それを調べるのがATAC-seq (アタックセック)です. トランポザーゼという酵素を用いて解けているDNAの配列のみを写し取ったものを断片化してから次世代シークエンサーで読む技術です.
RNA-seq
生体の様々なことを決める蛋白質, それはRNAから翻訳されることで作られます. よって細胞の中のRNAの量を測ることは生物学の研究ではほとんど避けては通れません.
ここでも次世代シークエンサーが大活躍します. 細胞からすべてのRNAを抽出して次世代シークエンサーでよんでしまえば, 細胞内にどのRNAがどれぐらいの量あるかを網羅的に調べることができます. これがRNA-seqです。
バイオインフォマティクス解析
いよいよバイオインフォマティクスについてお話します. バイオインフォマティクスとは次世代シークエンサーなどの技術により人力では到底扱いきれないほど大量に取得されたデータをコンピュータで解析する技術のことです.
マッピング
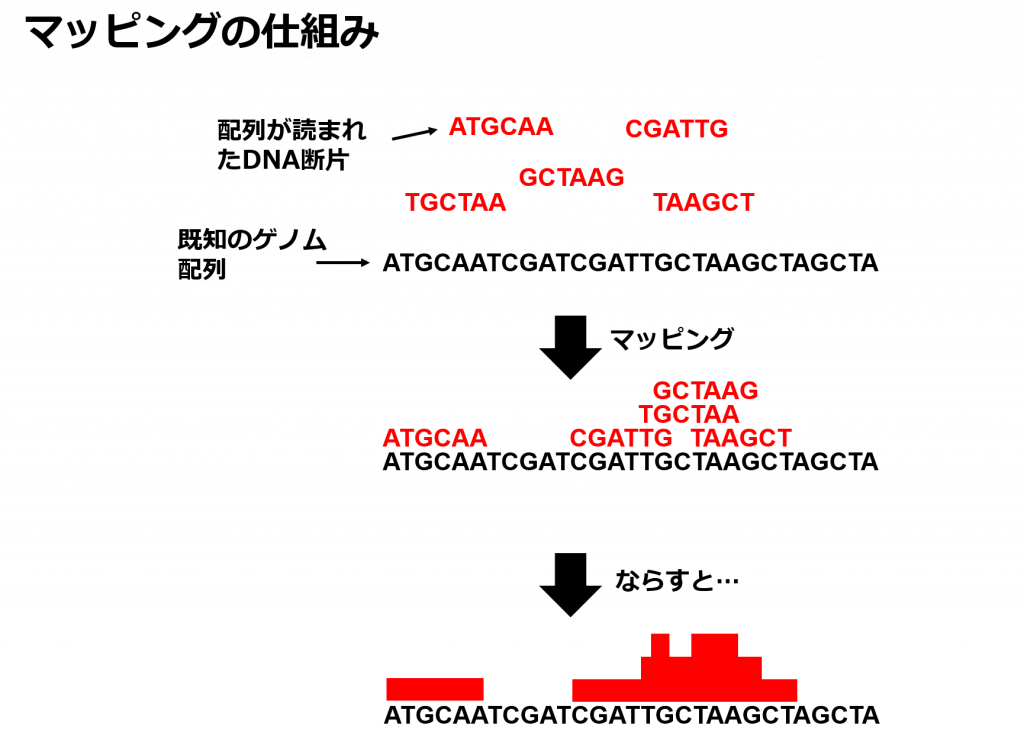
ここまで紹介した各種シークエンスから得られるのは短く断片化されたDNAの配列なので, その配列がゲノムのどの部分由来なのかを既知のゲノム配列と照らし合わせていく「マッピング」という作業を行わなければなりません.
RNA-seqも読むときにRNAをDNAに変換しています. なので同じようにマッピングすることができます.
短い断片といっても50~200塩基ほど長さがあります. さらにヒトゲノムは30億塩基からなるのでマッピングを手動でやると一生かかっても終わらないでしょう.
そこでコンピュータの出番です!コンピュータならマッピングをなんと10分ほどで行うことができます!コンピュータすごい!これで私がマッピングだけで人生を終えることはありません.
さてマッピングを行うと下図のように読まれたDNA断片の山ができます. この山の高さがChIP-seqではその領域にどれ位の確率で蛋白質が結合していたりヒストンマーカーがあるか, ATAC-seqではその領域がどれ位の確率で解けているか, RNA-seqではその領域から転写されるRNAが細胞中にどれぐらいの量存在するかを測ることができます.
大量のデータをコンピュータで解析する
無事マッピングを終えて, あなたは大量の遺伝子の情報を得ました.このデータをまとめるのもコンピュータの仕事であり, バイオインフォマティクスの醍醐味です.
ここではバイオインフォマティクスによるデータのまとめ方について紹介します.
遺伝子エンリッチメント解析
たとえば細胞を刺激したときのある転写因子AのChIP-seqのデータを取得したとします. つまりAがDNAのどこに結合しているかの情報を得たとします.
そのデータからAがどのような遺伝子の制御領域に結合しているかをコンピュータに計算させることができます. あなたはこれでAが制御している可能性のある遺伝子の名前のリストを手に入れました!5000個ぐらい!
5000個すべてググりますか?それは無理です. なんとかしてどのような機能を持つ遺伝子が多いのかなどの情報を得たいところです.
そこで遺伝子エンリッチメント解析です. 生物学は日進月歩, 毎日様々な遺伝子の機能に関する情報が報告されています. 世の中にはこのよう報告を登録したデータベースが多数存在します. 有名なものとしてGene Ontology (GO) や Kyoto Encyclopedia of Genes and Genomes (KEGG) などがあります.
遺伝子エンリッチメント解析では自分の遺伝子リストをデータベースに参照する計算を行うことでどのような機能を持つ遺伝子が多いか, どのような病気に関わる遺伝子が多いかなどの情報を引き出すことができます.
次元圧縮
がんという恐ろしい病気があります. がんの治療が難しい理由, それはがんは様々な状態の細胞の集まりなので一つの治療方法に対して抵抗できる細胞がたいてい存在するからです.
ではどのような細胞がいるのか. シークエンスで調べましょう!
現在の技術では1細胞に対してRNA-seqとATAC-seqを行うことができます. 患者さんからがん腫瘍を取ってきて細胞をたくさん取得してシークエンスをしてみましょう!
得られるデータはヒトの1細胞RNA-seqなら一つの細胞につき数万種類の遺伝子それぞれの発現量が得られるのは数万次元のデータになります.
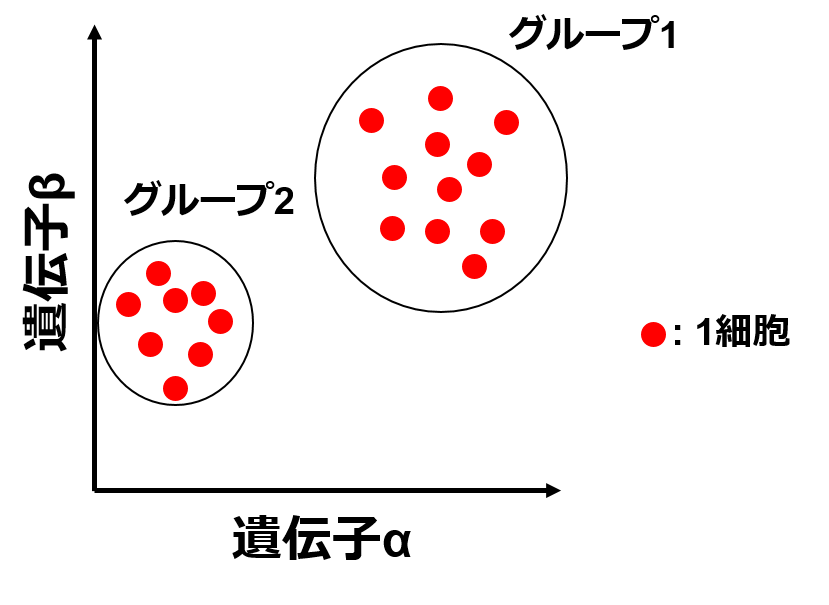
もし, 遺伝子が遺伝子α, βの二種類だけの二次元のデータだったなら細胞ごとの遺伝子αの発現量を横軸に, βの発現量を縦軸に取ってあげて点を打っていけば細胞集団の様子を二次元の平面に表して, 集団をグループ分けできるかもしれません.
ところが扱うデータは数万次元です, 人間は4次元以上の空間をとらえることはできません. どうすればよいでしょうか.
そこで次元圧縮という方法を使います. 詳しい説明は省きますが, イメージとしては三次元の立方体を二次元の絵に描くことができるように次元を落としていく作業を行って多次元のデータを二次元もしくは三次元のグラフに表します. このような方法にはPCA, t-SNE, UMAPといった方法があります.
以下の図のように細胞集団を遺伝子の発現量に基づいてグループ分けすることができます.

ここからいろいろな解析ができますが例えばそれぞれのグループの細胞でよく発現している遺伝子のリストを取得し, それを先程の遺伝子エンリッチメント解析にかけてみましょう.
すると各集団はどのような役割をもつ遺伝子がよく発現しているかがわかります. この情報からその細胞集団を攻撃するための薬の設計を行うことができるかもしれません.
また, 最近ではどの集団がどの集団に移り変わっているのかを予測することもできるようになってきているのでがんの発達の仕組みについて解明されることに繋がるかもしれません.
このようにバイオインフォマティクスのおかげでがんの仕組みの解明に人類はさらに近づいていっています.
ここではバイオインフォマティクス解析の一例として遺伝子エンリッチメント解析と次元圧縮の2つを紹介しました. シークエンスデータはとても巨大で実に多くの情報を得ることができるので他にももっとたくさんの解析できます. 興味がある方はぜひ調べてみてください.
きみもバイオインフォマティクスを始めよう!
シークエンスデータはネットから取り放題!
シークエンスデータを用いて研究を行って論文を投稿する際, ほとんどの場合データを公開することが義務となっています.
基本的には論文に開示先が載っていますし, データーベースで検索すると自分のほしいデータが見つかるかもしれません. 例えばChIP-seqのデータベースでは”ChIP-Atlas” ( https://chip-atlas.org/ )などがあります.
解析のやり方はネットで読み放題!
これはバイオインフォマティクスに限ったことではないのですがいまやネットでいろいろな情報が無料で手に入ります. 特にプログラミングなどのPCスキル系の情報は質のいい情報が日本語で溢れています.
バイオインフォマティクスにおいては”bioinformatics” ( https://bi.biopapyrus.jp/ ) という日本語のサイトがあり, とてもわかりやすく解析の仕方が説明されています. 私もこのサイトによくお世話になっています.
この他にもわかりやすいサイトや記事がネットには溢れているので検索してみてください.
データの解析には主に統計解析プログラミング言語であるRを使用することをオススメします. バイオインフォマティクスで最もよく用いられている言語で情報も豊富です.こちらも”R-Tips” ( http://cse.naro.affrc.go.jp/takezawa/r-tips/r.html ) など非常に親切なサイトがたくさんあります.
あとはみなさんのパソコンのOSがMacならばターミナルを開いてネットで調べながら解析を始めることができます. WindowsならばWSL (Windows Subsystem for Linux) をインストールして解析を始めてください.
さて, バイオインフォマティクスを含め情報解析を行うのにいちばん大事な力は「検索能力」です. いかにネットから自分がほしい情報を探し出すことができるか, これに尽きます.
ですからあとはみなさんの「検索能力」で調べまくってバイオインフォマティクスをエンジョイしてください!
参考文献
- Grosselin et al., High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer Nat. Genet. 51, 1060–1066 (2019)
- Whyte et al., Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes Cell 153, 307–319 (2013)
- Ashburner et al., Gene ontology: tool for the unification of biology Nat. Genet. 25, 25-29 (2000)
- Kanehisa et al., KEGG: Kyoto Encyclopedia of Genes and Genome Nucl Acid Res 28, 27-30 (2000)
- Patel et al., Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma Science 344, 1396-1401 (2014)